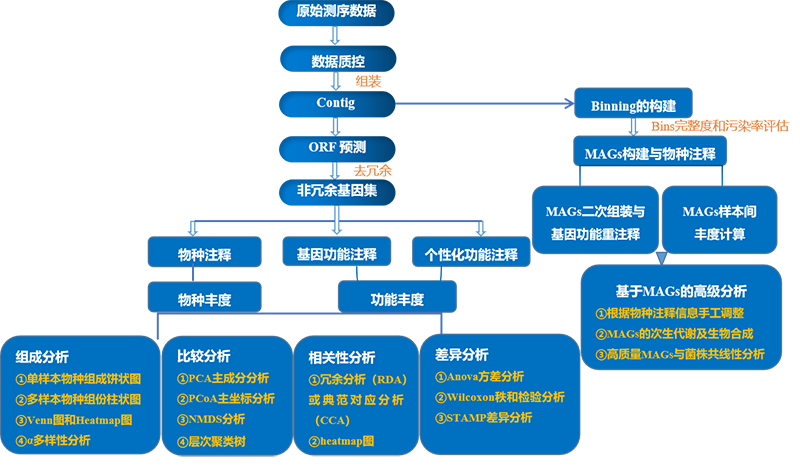
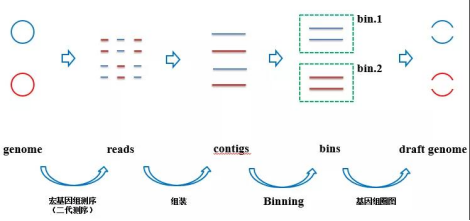
自然环境样品中存在成百上千种微生物,获得这些微生物的物种信息和功能信息是宏基因组研究的重点和难点。常规的分析手段都是在基因层面去获得的物种组成和功能分布,是所有微生物的总和。如果想要从环境样品中获得一些菌株的基因组信息,就需要借助宏基因组Binning和HiC-Meta等辅助手段,通过这些技术手段,可以得到一些菌株的框架图。
HiC-Meta是针对环境样品,研究整个染色质DNA在空间位置上的关系,获得高分辨率染色质三维结构信息,该技术需要构建Hi-C文库,然后配套生物信息学分析,价格昂贵,一般课题组很难接受大规模Hi-C样品的实验。相对而言,宏基因组Binning门槛就低很多,只需要在现有基础上,加大测序深度(推荐测20-50G),即可用生物信息学的手段,获得高质量的菌株框架图。
Binning即分箱、聚类,指从微生物群体序列中将不同个体的序列(reads或contigs等)分离开来的过程。简单来说,就是把宏基因组数据中来自同一菌株的序列聚到一起,得到一个菌株的基因组。
Binning类型
|
序列类型 |
优势 |
原因 |
|
Reads Binning |
可聚类出宏基因组中丰度非常低的物种 |
宏基因组组装中reads利用率低,单样品5Gb测序量情况下,环境样品组装reads利用率一般只有10%左右,肠道样品或极端环境样品组装reads利用率一般能达到30%,低丰度的物种可能没有被组装出来,没有体现在gene或者contig中,因此基于reads Binning才有可能得到低丰度的物种。 |
|
Contigs Binning |
效果更好 |
核酸组成和物种丰度变化模式在越长的序列中越显著和稳定。 |
|
Genes Binning |
应用广泛 |
宏基因组分析中都会计算gene丰度,基于genes丰度变化模式进行Binning,可操作性较强;基于genes Binning有很多可参考的文献,过程也并不复杂,可复制性强,对计算机资源消耗也较低。 |
不依赖于微生物的分离培养,是环境微生物单菌基因组(框架图)研究的一种新途径和高性价比策略;可得到环境中丰度较低的微生物,为研究低丰度微生物提供了新途径;引入了宏观生态的研究理念,对环境中微生物菌群的多样性、功能活性等宏观特征进行研究,更准确地反映出微生物的真实状态;多组学关联分析,可关联代谢组等研究结果,全方面深入研究。